MetaVolcanoR: Differential expression meta-analysis tool
Feb 1, 2026
Source:vignettes/MetaVolcano.Rmd
MetaVolcano.RmdIntroduction: Why Meta-Analysis?
Imagine you’re studying a disease and find 5 different studies on Gene Expression Omnibus, each identifying ~500 differentially expressed genes (DEGs). But only 50 genes overlap! Which genes are truly important?
MetaVolcanoR solves this by: - Combining evidence across studies - Identifying consistently perturbed genes - Visualizing meta-analysis results intuitively
Comparing the expression of genes under a given condition against a reference biological state is usually applied to identify sets of differentially expressed genes (DEG). These DEG point out the genomic regions functionally relevant under the biological condition of interest.
Athough individual genome-wide expression studies have small signal/noise ratio, today’s genomic data availability usually allows to combine differential gene expression results from dozens of independent studies to overcome this limitation.
Databases such as GEO (https://www.ncbi.nlm.nih.gov/geo/), SRA (https://www.ncbi.nlm.nih.gov/sra), ArrayExpress, (https://www.ebi.ac.uk/arrayexpress/), and ENA (https://www.ebi.ac.uk/ena) offer systematic access to vast amounts of transcriptome data. There exists more than one gene expression study for many biological conditions. This redundancy could be exploit by meta-analysis approaches to reveal genes that are consistently present and differentially expressed under given conditions.
MetaVolcanoR was designed to identify the genes whose expression is consistently perturbed across several DE tables.
Usage
Overview
The MetaVolcanoR R package combines differential gene expression results. It implements three strategies to summarize gene expression activities from different studies. i) Random Effects Model (REM) approach. ii) a vote-counting approach, and iii) a p-value combining-approach. MetaVolcano exploits the Volcano plot reasoning to visualize these meta-analysis of gene expression results.
Preparing Your Data
MetaVolcanoR requires differential expression results with:
- Gene identifiers (gene names or IDs)
- Log2 fold changes
- P-values
- Confidence intervals OR variance (for REM method only)
If you have confidence intervals (CI)
If your DE results include confidence interval columns (example:
CI.L and CI.R), you're ready to go! The
package will automatically calculate variance:
# Your data has CI.L and CI.R columns
meta_rem <- rem_mv(
diffexp = your_data_list,
llcol = "CI.L", # Left limit of CI
rlcol = "CI.R", # Right limit of CI
cvar = TRUE # Calculate variance from CI (default)
)If you have standard error (SE)
Most tools (DESeq2, limma, edgeR) output standard error, not confidence intervals. Convert SE to 95% CI:
# From DESeq2 results
deseq_results <- results(dds) #or after shrinkage
deseq_results$CI.L <- deseq_results$log2FoldChange - 1.96 * deseq_results$lfcSE
deseq_results$CI.R <- deseq_results$log2FoldChange + 1.96 * deseq_results$lfcSE
# From limma results (if you have SE column)
limma_results$CI.L <- limma_results$logFC - 1.96 * limma_results$SE
limma_results$CI.R <- limma_results$logFC + 1.96 * limma_results$SEIf you have variance directly
If you have variance (or can calculate it from SE:
variance = SE^2), use the vcol parameter:
# Calculate variance from standard error
your_data$variance <- your_data$SE^2
# Use variance directly
meta_rem <- rem_mv(
diffexp = your_data_list,
vcol = "variance", # Column name with variance
cvar = FALSE # Don\'t calculate from CI
)Estimating variance from test statistics
If your differential expression results do not include confidence
interval columns, variance can be approximated from the fold-change and
test statistic (e.g. z-score or t-statistic), using the relationship
stat ≈ log2FC / SE, which gives
var = (log2FC / stat)^2.
If your data is already organized as a named list of data frames, you can add the variance column as follows:
# If your tables are already in a list called diffexp_list:
diffexp_list <- lapply(diffexp_list, function(df) {
df %>%
mutate(var = ifelse(stat == 0 | is.na(stat),
NA_real_,
(log2FC / stat)^2))
})Note the ifelse guard — transcripts where
stat = 0 would produce infinite variance and cause
rem_mv to fail, so these are set to NA and
will be handled gracefully by the model (flagged as
error = TRUE).
Then pass vcol = "var" and cvar = FALSE to
rem_mv:
meta_mv <- rem_mv(
diffexp = diffexp_list,
pcriteria = "pvalue",
foldchangecol = "log2FC",
genenamecol = "Symbol",
geneidcol = "Symbol",
llcol = NULL,
rlcol = NULL,
vcol = "var",
cvar = FALSE,
...
)Preparing Swish/fishpond transcript-level results
If your differential expression analysis was performed at the
transcript level using swish() from the fishpond package,
MetaVolcanoR can integrate your results using an approximate estimate of
variance derived from Swish’s test statistic. This lets you go straight
from the differential transcript expression (DET) results table to a
MetaVolcanoR-ready format, without needing to carry the
SummarizedExperiment object or its inferential replicates
forward.
Checking your DET table
Before preparing your data, inspect the columns available in your results table:
colnames(det)swish() adds the following statistical result columns to
rowData(): stat, log2FC,
pvalue, locfdr, qvalue. If you
extracted your DET table with something like
det <- as.data.frame(mcols(se)), these columns carry
over directly. Identifier columns (e.g. tx_id,
tx_name) depend on how you imported your quantification
data — if you used tximeta::tximeta(), these are typically
added automatically based on the salmon index and usually contain
Ensembl transcript IDs (e.g. ENST00000456328.2) rather than
human-readable isoform names (e.g. HES4-202).
prepare_swish() auto-detects the identifier column by
looking for tx_name, tx_id,
transcript_id, or transcript_name (in that
order) among your column names — no need to pass tx_col if
one of these is present. If your identifier column has a different name,
or if you plan to combine several studies with rem_mv(),
pass tx_col explicitly and make sure the identifiers are
consistent across all studies (same ID type, same
versioning). Any ID harmonization should be done upstream, on your DET
table, prior to this step.
Preparing swish results for MetaVolcanoR
Once your DET table has a consistent identifier column, use
prepare_swish() to generate the table required by
MetaVolcanoR:
# Auto-detects the identifier column (tx_name, tx_id, etc.)
det_table <- prepare_swish(det = det_1)
# Or specify explicitly if needed
det_table <- prepare_swish(det = det_1, tx_col = "tx_name")If your identifiers are plain Ensembl transcript IDs, the
Symbol column will simply contain values like
ENST00000456328.2. The meta-analysis will still run without
any issue — this only affects how gene/transcript labels look on the
volcano plot:
det_table <- prepare_swish(det = det_1, tx_col = "tx_id")Complete workflow with
prepare_swish()
library(fishpond)
library(SummarizedExperiment)
library(MetaVolcanoR)
# Extract DET tables from your swish results
det_1 <- as.data.frame(mcols(se_swish_1))
det_2 <- as.data.frame(mcols(se_swish_2))
# Prepare each study
study_1 <- prepare_swish(det = det_1, tx_col = "tx_name")
study_2 <- prepare_swish(det = det_2, tx_col = "tx_name")
# Combine into named list — names are required
my_studies <- list(
study1 = study_1,
study2 = study_2
)
# Run REM meta-analysis
meta_results <- rem_mv(
diffexp = my_studies,
pcriteria = "pvalue",
foldchangecol = "Log2FC",
genenamecol = "Symbol",
llcol = "CI.L",
rlcol = "CI.R",
cvar = TRUE,
metathr = 0.01,
draw = "HTML"
)
meta_results@MetaVolcanoPreparing reuslts from common DE tools
MetaVolcanoR provides convenient helper functions to prepare results from common DE tools:
# For DESeq2 results
deg_table <- prepare_deseq2(res)
# For limma results
deg_table <- prepare_limma(limma_toptable)
# For edgeR results
deg_table <- prepare_edger(edger_toptags$table)
# For Swish/fishpond results (transcript-level)
det_table <- prepare_swish(det)Complete workflow with helper functions:
library(DESeq2)
library(MetaVolcanoR)
# Run your DESeq2 analysis
dds <- DESeqDataSetFromMatrix(count_matrix, sample_info, design = ~ condition)
dds <- DESeq(dds)
# Get results for multiple comparisons
res_treatment1 <- results(dds, contrast = c("condition", "Treatment1", "Control"))
res_treatment2 <- results(dds, contrast = c("condition", "Treatment2", "Control"))
res_treatment3 <- results(dds, contrast = c("condition", "Treatment3", "Control"))
# Prepare all studies using helper function
study1 <- prepare_deseq2(res_treatment1)
study2 <- prepare_deseq2(res_treatment2)
study3 <- prepare_deseq2(res_treatment3)
# Combine into named list
my_studies <- list(
"Treatment1_vs_Control" = study1,
"Treatment2_vs_Control" = study2,
"Treatment3_vs_Control" = study3
)
# Run meta-analysis
meta_results <- rem_mv(
diffexp = my_studies,
metathr = 0.01,
outputfolder = tempdir(),
draw = "HTML"
)
# View results
meta_results@MetaVolcano
head(meta_results@metaresult)Testing the helper functions:
You can test if your data is correctly formatted:
## Symbol Log2FC pvalue CI.L CI.R
## 1 A1BG -0.70126879 0.000140100 -1.0087857 -0.39375189
## 2 A1BG-AS1 -0.25106351 0.008694757 -0.4304790 -0.07164803
## 3 A1CF 0.03332573 0.615989488 -0.1036882 0.17033968
## 4 A2M 0.83504214 0.018550388 0.1568214 1.51326289
## 5 A2ML1 0.03942552 0.843222358 -0.3728473 0.45169836
## 6 A4GALT -0.20815882 0.282488068 -0.6025247 0.18620708
# Your prepared data should have these columns:
# - Symbol (or gene identifier)
# - Log2FC (fold change)
# - pvalue (p-value)
# - CI.L (lower confidence interval, for REM only)
# - CI.R (upper confidence interval, for REM only)
# Test with one study
str(diffexplist[[1]])## 'data.frame': 6573 obs. of 5 variables:
## $ Symbol: chr "A1BG" "A1BG-AS1" "A1CF" "A2M" ...
## $ Log2FC: num -0.7013 -0.2511 0.0333 0.835 0.0394 ...
## $ pvalue: num 0.00014 0.00869 0.61599 0.01855 0.84322 ...
## $ CI.L : num -1.009 -0.43 -0.104 0.157 -0.373 ...
## $ CI.R : num -0.3938 -0.0716 0.1703 1.5133 0.4517 ...
## - attr(*, ".internal.selfref")=<externalptr>Note: The prepare_* functions
automatically: - Remove rows with NA values - Calculate 95% confidence
intervals from standard errors - Format column names to match
MetaVolcanoR requirements - Filter out infinite values
Implemented meta-analysis approaches
Random Effect Model MetaVolcano
The REM MetaVolcano summarizes the gene fold change of several studies taking into account the variance. The REM estimates a summary p-value which stands for the probability of the summary fold-change is not different than zero. Users can set the metathr parameter to highlight the top percentage of the most consistently perturbed genes. This perturbation ranking is defined following the topconfects approach.
meta_degs_rem <- rem_mv(diffexp=diffexplist,
pcriteria="pvalue",
foldchangecol='Log2FC',
genenamecol='Symbol',
geneidcol=NULL,
collaps=FALSE,
llcol='CI.L',
rlcol='CI.R',
vcol=NULL,
cvar=TRUE,
metathr=0.01,
jobname="MetaVolcano",
outputfolder=".",
draw='HTML',
ncores=1)## index Symbol Log2FC_1 CI.L_1 CI.R_1 vi_1 Log2FC_2
## 1 4795 MXRA5 0.8150851 0.3109324 1.3192377 0.06616251 1.3001104
## 2 2166 COL6A6 -1.7480348 -2.5780749 -0.9179947 0.17934364 -0.8388366
## 3 2053 CIDEA NA NA NA NA NA
## 4 7115 SULT1A4 0.9689025 0.5103475 1.4274575 0.05473571 0.7513323
## 5 130 ACACB -0.8431142 -1.4708480 -0.2153804 0.10257437 -1.1119841
## 6 6528 SLC27A2 -0.6782948 -0.9931027 -0.3634869 0.02579759 -1.8916655
## CI.L_2 CI.R_2 vi_2 Log2FC_3 CI.L_3 CI.R_3 vi_3
## 1 0.6603306 1.9398901 0.10654886 1.1895480 0.8401301 1.5389659 0.031781777
## 2 -1.3578456 -0.3198277 0.07011930 -1.0300519 -1.4730328 -0.5870710 0.051080819
## 3 NA NA NA -1.0111528 -1.3226326 -0.6996729 0.025255027
## 4 0.4707021 1.0319624 0.02050012 NA NA NA NA
## 5 -1.7417389 -0.4822293 0.10323592 -0.5305046 -0.6957455 -0.3652637 0.007107599
## 6 -2.6822584 -1.1010726 0.16270229 -1.2126830 -1.6702908 -0.7550753 0.054509799
## Log2FC_4 CI.L_4 CI.R_4 vi_4 Log2FC_5 CI.L_5 CI.R_5
## 1 0.2188594 -1.052230 1.4899492 0.4205720 0.8051543 0.1367255 1.4735830
## 2 -1.3755263 -2.162453 -0.5885999 0.1611967 -0.7213490 -1.5714484 0.1287505
## 3 -1.7991026 -2.918939 -0.6792665 0.3264351 -0.8738120 -1.6373061 -0.1103179
## 4 NA NA NA NA NA NA NA
## 5 -0.7991042 -1.457868 -0.1403403 0.1129659 -0.5155929 -0.8606782 -0.1705076
## 6 -1.3554403 -2.288444 -0.4224370 0.2265970 -1.4905464 -2.5565023 -0.4245905
## vi_5 signcon ntimes randomSummary randomCi.lb randomCi.ub randomP
## 1 0.11630493 5 5 1.0333001 0.7882044 1.2783958 1.420312e-16
## 2 0.18811668 -5 5 -1.0649749 -1.3396138 -0.7903361 2.956522e-14
## 3 0.15173972 -3 3 -1.0417876 -1.3210774 -0.7624977 2.653168e-13
## 4 NA 2 2 0.8106154 0.5712566 1.0499741 3.187477e-11
## 5 0.03099851 -5 5 -0.5830624 -0.7212245 -0.4449003 1.324963e-16
## 6 0.29577833 -5 5 -1.2207058 -1.6760435 -0.7653680 1.484852e-07
## het_QE het_QEp het_QM het_QMp error se rank
## 1 4.179945 0.38220032 68.27752 1.420312e-16 FALSE 0.12504883 1
## 2 4.580708 0.33308457 57.76318 2.956522e-14 FALSE 0.14012185 2
## 3 1.980047 0.37156797 53.44957 2.653168e-13 FALSE 0.14249482 3
## 4 0.629179 0.42765661 44.05825 3.187477e-11 FALSE 0.12212181 4
## 5 4.317851 0.36469506 68.41455 1.324963e-16 FALSE 0.07049086 5
## 6 11.099093 0.02547263 27.60901 1.484852e-07 FALSE 0.23231516 6
head(meta_degs_rem@metaresult, 3)## Symbol signcon randomSummary randomCi.lb randomCi.ub randomP het_QE
## 1 MXRA5 5 1.033300 0.7882044 1.2783958 1.420312e-16 4.179945
## 2 COL6A6 -5 -1.064975 -1.3396138 -0.7903361 2.956522e-14 4.580708
## 3 CIDEA -3 -1.041788 -1.3210774 -0.7624977 2.653168e-13 1.980047
## het_QEp het_QM het_QMp error rank
## 1 0.3822003 68.27752 1.420312e-16 FALSE 1
## 2 0.3330846 57.76318 2.956522e-14 FALSE 2
## 3 0.3715680 53.44957 2.653168e-13 FALSE 3
meta_degs_rem@MetaVolcano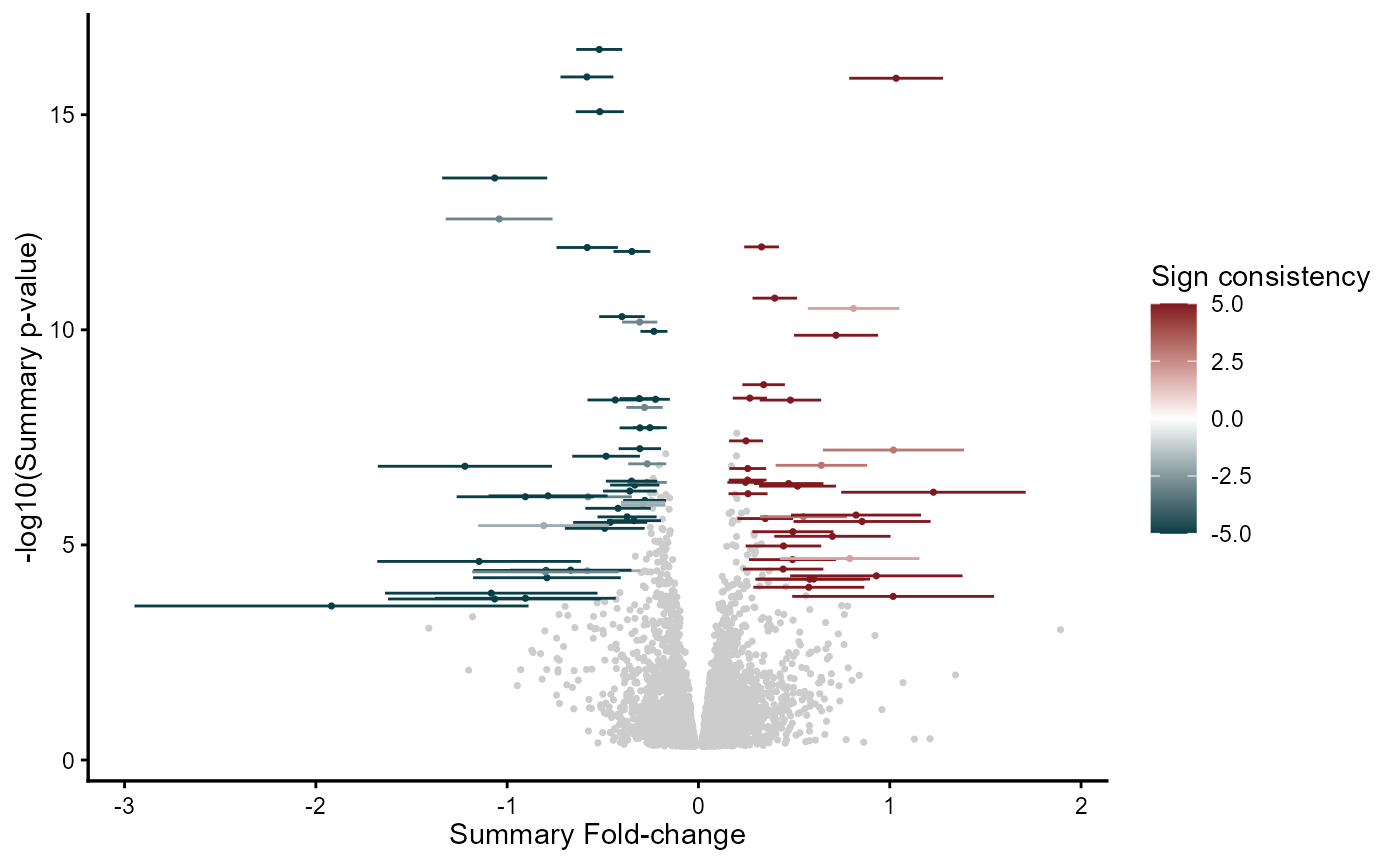
draw_forest(remres=meta_degs_rem,
gene="MMP9",
genecol="Symbol",
foldchangecol="Log2FC",
llcol="CI.L",
rlcol="CI.R",
jobname="MetaVolcano",
outputfolder=".",
draw="HTML")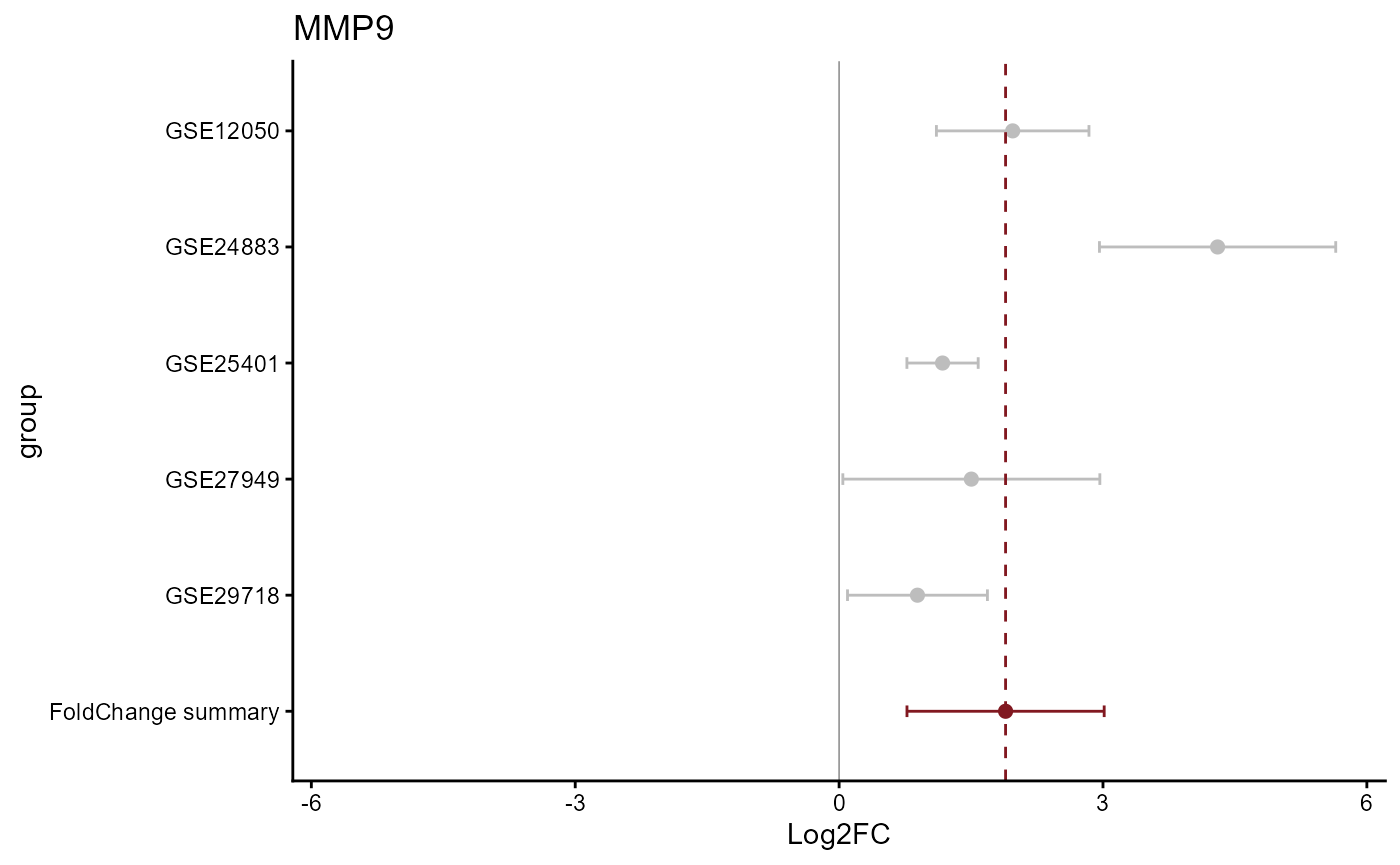
draw_forest(remres=meta_degs_rem,
gene="COL6A6",
genecol="Symbol",
foldchangecol="Log2FC",
llcol="CI.L",
rlcol="CI.R",
jobname="MetaVolcano",
outputfolder=".",
draw="HTML")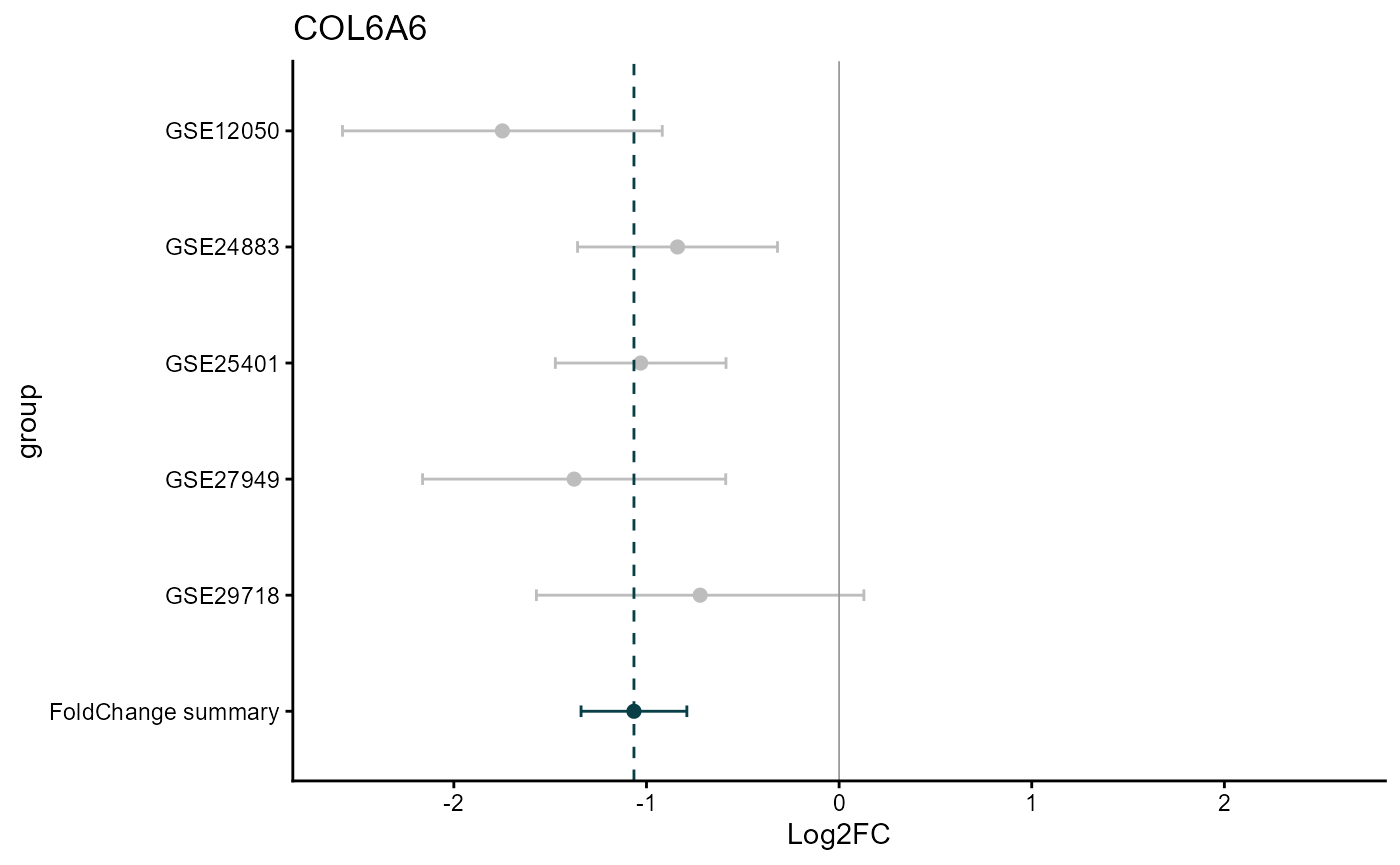
The REM MetaVolcano also allows users to explore the forest plot of a given gene based on the REM results.
Vote-counting approach
The vote-counting MetaVolcano identifies differential expressed genes (DEG) for each study based on the user-defined p-value and fold change thresholds. It displays the number of differentially expressed and unperturbed genes per study. In addition, it plots the inverse cumulative distribution of the consistently DEG, so the user can identify the number of genes whose expression is perturbed in at least 1 or n studies.
meta_degs_vote <- votecount_mv(diffexp=diffexplist,
pcriteria='pvalue',
foldchangecol='Log2FC',
genenamecol='Symbol',
geneidcol=NULL,
pvalue=0.05,
foldchange=0,
metathr=0.01,
collaps=FALSE,
jobname="MetaVolcano",
outputfolder=".",
draw='HTML')
head(meta_degs_vote@metaresult, 3)## Symbol deg_1 deg_2 deg_3 deg_4 deg_5 ndeg ddeg idx degvcount
## 1 ABCC3 1 1 1 1 1 5 5 25 2.Up-regulated
## 2 ABHD5 -1 -1 -1 -1 -1 5 -5 -25 0.Down-regulated
## 3 ACACB -1 -1 -1 -1 -1 5 -5 -25 0.Down-regulated
meta_degs_vote@featurefreq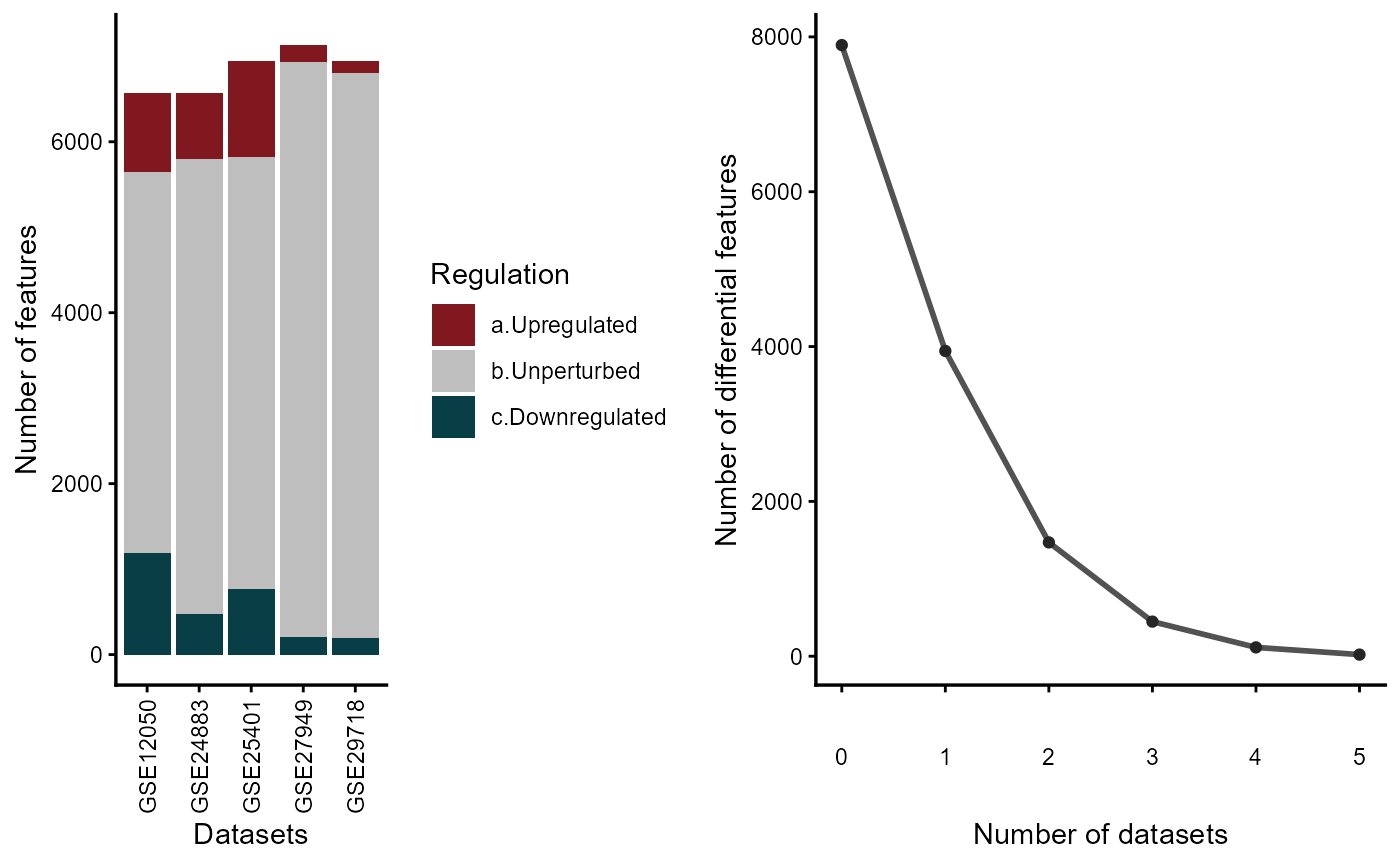
The vote-counting MetaVolcano visualizes genes based on the number of studies where genes were identified as differentially expressed and the gene fold change sign consistency. It means that a gene that was differentially expressed in five studies, from which three of them it was downregulated, will get a sign consistency score of 2 + (-3) = -1. Based on user preference, MetaVolcano can highlight the top metathr percentage of consistently perturbed genes.
meta_degs_vote@MetaVolcano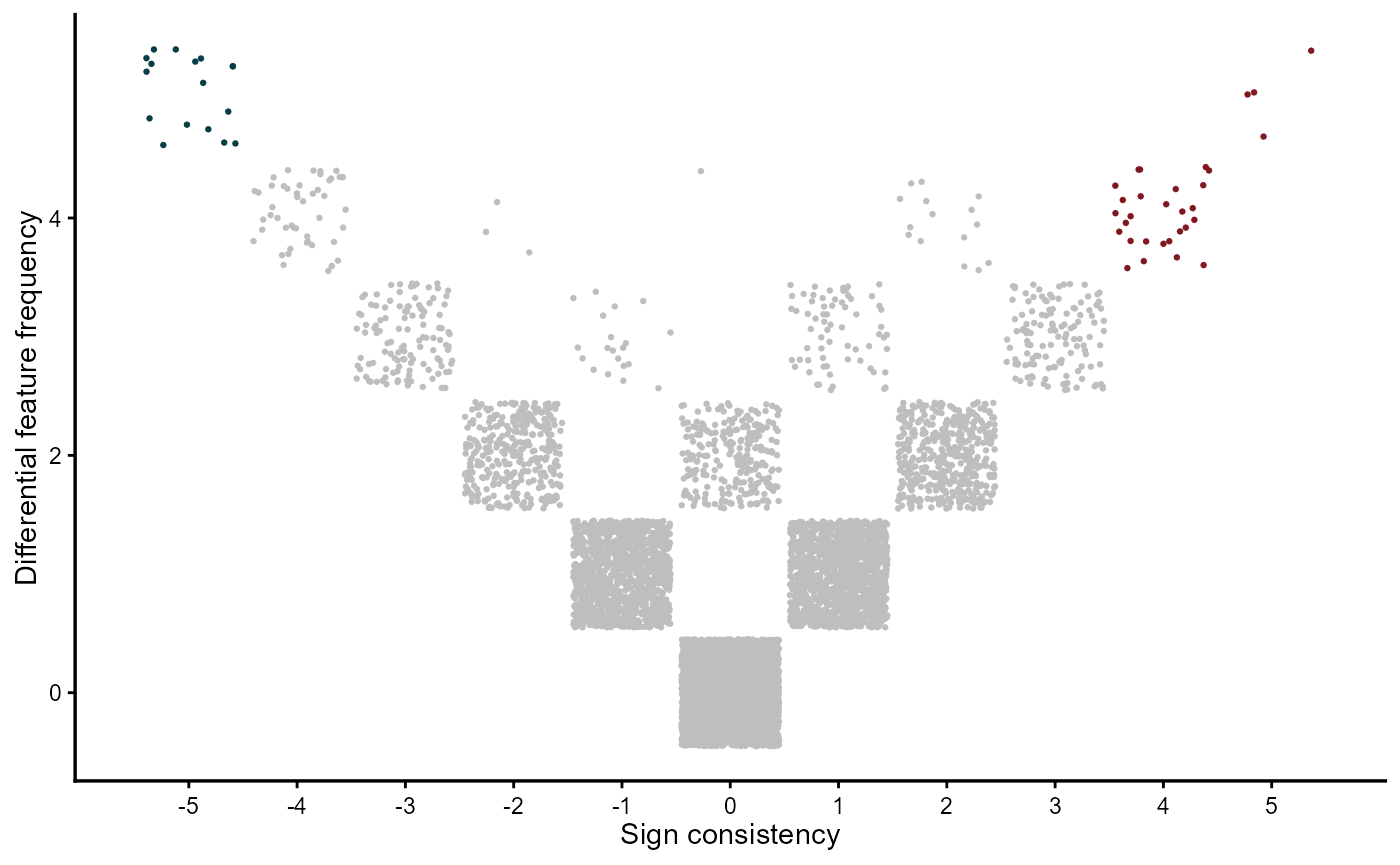
Combining-approach
The combinig MetaVolcano summarizes the fold change of a gene in different studies by the mean or median depending on the user preference. In addition, the combinig MetaVolcano summarizes the gene differential expression p-values using the Fisher method. The combining MetaVolcano can highlight the top metathr percentage of consistently perturbed genes.
meta_degs_comb <- combining_mv(diffexp=diffexplist,
pcriteria='pvalue',
foldchangecol='Log2FC',
genenamecol='Symbol',
geneidcol=NULL,
metafc='Mean',
metathr=0.01,
collaps=TRUE,
jobname="MetaVolcano",
outputfolder=".",
draw='HTML')
head(meta_degs_comb@metaresult, 3)## Symbol metap metafc idx
## 1 MMP9 9.002947e-15 1.9693517 27.66076
## 2 ACVR1C 3.548802e-20 -1.2544105 -24.39818
## 3 ANG 5.674270e-26 -0.9364936 -23.64280
meta_degs_comb@MetaVolcano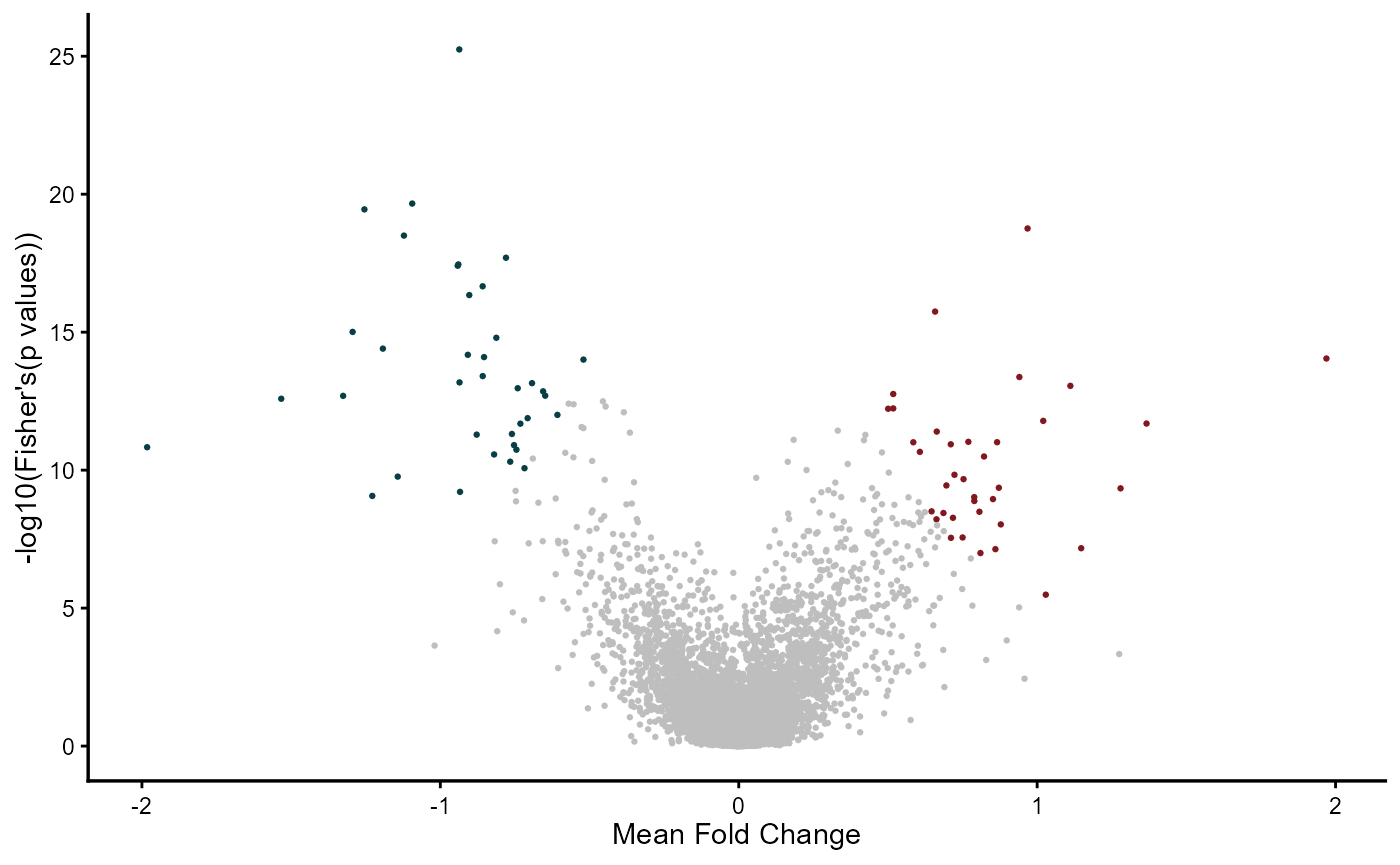
Customizing Your Plots
MetaVolcanoR provides extensive customization options to create publication-ready figures and highlight specific genes of interest.
Basic Customization Options
All main functions (rem_mv, votecount_mv,
combining_mv) support these parameters:
- colors: Custom color schemes
- point_size: Size of data points
- label_genes: Vector of specific genes to label
- label_top_n: Automatically label top N genes
- label_size: Size of gene labels
- plot_title: Custom plot title
- show_legend: Show or hide legend
Example 1: Custom Colors and Gene Labels
# REM with custom colors and specific genes labeled
meta_custom <- rem_mv(
diffexp = diffexplist,
metathr = 0.01,
outputfolder = tempdir(),
draw = "HTML",
# Customization parameters:
colors = c(low = "navy", mid = "white", high = "darkred", na = "gray80"),
point_size = 1.5,
label_genes = c("MMP9", "COL6A6", "MXRA5", "CIDEA"),
label_size = 4,
plot_title = "REM Meta-Analysis - Custom Colors",
show_legend = TRUE
)
meta_custom@MetaVolcanoExample 2: Highlighting Top Genes Automatically
# Vote-counting with automatic labeling of top 10 genes
meta_vote_labeled <- votecount_mv(
diffexp = diffexplist,
pvalue = 0.05,
metathr = 0.01,
outputfolder = tempdir(),
draw = "HTML",
# Automatically label top 10 most significant genes
colors = c("steelblue", "gray90", "firebrick"),
point_size = 1.2,
label_top_n = 10,
label_size = 3.5,
plot_title = "Vote-Counting: Top 10 DEGs Labeled"
)
meta_vote_labeled@MetaVolcanoExample 3: Publication-Ready Figures
# Combining approach with publication styling
meta_publication <- combining_mv(
diffexp = diffexplist,
metafc = "Median",
metathr = 0.01,
collaps = TRUE,
outputfolder = tempdir(),
draw = "HTML",
# Publication-ready styling
colors = c("darkgreen", "white", "darkorange"),
point_size = 1.0,
label_genes = c("MMP9", "ANG", "ACVR1C"),
label_size = 3,
plot_title = NULL, # No title for publication
show_legend = FALSE
)
meta_publication@MetaVolcanoExample 4: Customizing Forest Plots
Forest plots also support extensive customization:
# Custom forest plot with specific colors and dimensions
draw_forest(
remres = meta_degs_rem,
gene = "MMP9",
outputfolder = tempdir(),
draw = "PDF",
# Customization:
colors = c(
positive = "darkred", # Color for positive fold changes
negative = "steelblue", # Color for negative fold changes
neutral = "gray70", # Color for individual studies
reference = "black" # Color for reference lines
),
point_size = 3,
plot_width = 7, # Width in inches (for PDF)
plot_height = 6, # Height in inches
plot_title = "MMP9 Expression Meta-Analysis"
)Color Scheme Examples
Color Schemes for Different Contexts
# Professional/Conservative
colors_professional <- c(low = "blue", mid = "white", high = "red", na = "gray80")
# High Contrast (for presentations)
colors_presentation <- c(low = "purple", mid = "white", high = "orange", na = "lightgray")
# Color-blind friendly
colors_colorblind <- c(low = "#0072B2", mid = "white", high = "#D55E00", na = "gray80")
# Grayscale (for print)
colors_grayscale <- c(low = "black", mid = "gray90", high = "gray30", na = "gray70")
# Use any of these:
meta_rem <- rem_mv(
diffexp = diffexplist,
colors = colors_colorblind, # Or any other scheme
# ... other parameters
)Vote-counting and Combining Colors
For vote-counting and combining approaches, provide a vector of 3 colors:
# Custom colors: [downregulated, neutral, upregulated]
custom_colors <- c("navyblue", "gray85", "darkred")
meta_vote <- votecount_mv(
diffexp = diffexplist,
colors = custom_colors,
# ... other parameters
)Combining Multiple Customizations
# Complete customization example
meta_final <- rem_mv(
diffexp = diffexplist,
metathr = 0.01,
outputfolder = tempdir(),
draw = "HTML",
ncores = 4,
# All customization options
colors = c(low = "#0072B2", mid = "white", high = "#D55E00", na = "gray80"),
point_size = 1.5,
label_genes = c("MMP9", "COL6A6"), # Label specific genes
label_top_n = 5, # Also label top 5
label_size = 3.5,
plot_title = "Meta-Analysis: Disease vs Control",
show_legend = TRUE
)
# Access the customized plot
meta_final@MetaVolcano
# View the forest plot for a specific gene
draw_forest(
remres = meta_final,
gene = "MMP9",
outputfolder = tempdir(),
draw = "PDF",
colors = c(positive = "#D55E00", negative = "#0072B2",
neutral = "gray60", reference = "black"),
point_size = 3,
plot_width = 8,
plot_height = 6
)Tips for Publication-Quality Figures
- Use color-blind friendly palettes (see examples above)
-
Set
show_legend = FALSEfor cleaner figures (add legend in figure caption) - Label only key genes instead of showing all labels
-
Use
plot_title = NULLand add titles in your manuscript -
Save as PDF (
draw = "PDF") for vector graphics in publications -
Adjust
plot_widthandplot_heightto match journal requirements - Use consistent colors across all figures in your paper
Saving Plots Separately
If you want to save plots with specific dimensions without displaying them:
# Generate the meta-analysis
meta_result <- rem_mv(
diffexp = diffexplist,
draw = "HTML", # or "PDF"
outputfolder = "path/to/output",
# ... other parameters
)
# The plot is automatically saved to the outputfolder
# For HTML: interactive plot you can explore in browser
# For PDF: publication-ready vector graphics